Software hilft Embryonalentwicklung zu entschlüsseln
Wissenschaftler aus Tübingen entwickeln Software zur Simulation von Netzwerken, die den Ablauf der Embryonalentwicklung steuern
Wenn neues Leben entsteht, wird aus einem winzigen Haufen identischer Zellen ein ausgewachsener Organismus mit verschiedenen Körperteilen. Vor 60 Jahren schlug Alan Turing die Theorie vor, dass solche Musterbildungsvorgänge während der Embryogenese durch die Ausbreitung zweier unterschiedlicher Signalmoleküle in den sich entwickelnden Geweben gesteuert werden. Wissenschaftler vom Friedrich-Miescher-Laboratorium der Max-Planck-Gesellschaft in Tübingen haben jetzt eine Software entwickelt, die mittels eines mathematischen Algorithmus systematisch realistische Musterbildungssysteme mit mehr als zwei Molekülen analysieren kann. Mit der Software haben die Forscher Musterbildungsprozesse während der Embryonalentwicklung untersucht. Sie können nun neue Systeme für den Einsatz in der Biotechnologie und regenerativen Medizin aufzeigen.

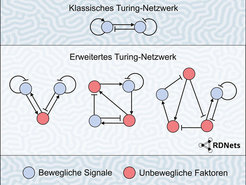
Vor mehr als sechs Jahrzehnten postulierte Alan Turing - vielen bekannt als Vater der modernen Computertechnologie und berühmt für die Entschlüsselung der Enigma-Nachrichten während des zweiten Weltkriegs - ein Modell der Embryonalentwicklung. In Turings sogenanntem Reaktions-Diffusions-Modell reagieren zwei Arten von Signalmolekülen miteinander, die sich durch Diffusion im Embryo ausbreiten. Turing bewies mathematisch, dass die beiden Signalmoleküle räumliche Muster bilden können, wenn sich eines der Moleküle schneller als das andere bewegt. An verschiedenen Positionen im Embryo entstehen so Muster unterschiedlich hoher Molekülkonzentrationen, die den Zellen die Informationen liefern, wo verschiedene Körperteile angelegt werden sollen. Obwohl Turings Modelle den realen Mustern während der Embryonalentwicklung sehr nahe kommen, war sein Ansatz auf zwei Signalmoleküle beschränkt. Tatsächlich liegen der Musterbildung während der Embryonalentwicklung aber komplexe genregulatorische Netzwerke mit mehr als zwei Molekülen zugrunde, die jedoch erst in den Jahrzehnten nach Turings Tod entdeckt wurden.
Jetzt hebt eine Arbeit Turings Reaktions-Diffusions-Modell in das Zeitalter der Postgenomik. Ein Team um Patrick Müller vom Friedrich-Miescher-Laboratorium der Max-Planck-Gesellschaft und Wissenschaftler des Centre for Genomic Regulation in Barcelona haben eine neue computergestützte Methode entwickelt, mit der die Musterbildung von Reaktions-Diffusions-Netzwerken systematisch analysiert und simuliert werden kann. Der neue Ansatz berücksichtigt bei den zugrundeliegenden genregulatorischen Netzwerken neben beweglichen Signalmolekülen auch unbewegliche Faktoren, wie etwa Rezeptoren für die Signalmoleküle auf den Zellen des Gewebes. „Musterbildende Systeme im wirklichen Leben bestehen nicht aus den vereinfachten Zwei-Komponenten-Netzwerken der klassischen Turing-Modelle. Wir wollten komplexere Systeme untersuchen und haben eine anwenderfreundliche Software entwickelt, mit der wir biologisch relevante Netzwerke entdecken können“, sagt Luciano Marcon, Erstautor der Veröffentlichung.
Software beschleunigt Berechnungen
Die Analyse von Turing-Modellen erfordert langwierige mathematische Berechnungen, die typischerweise von Hand durchgeführt werden und zwei Seiten füllen würden, um ein einziges Netzwerk zu modellieren. Diesen Ansatz so auszuweiten, dass Millionen möglicher Netzwerke systematisch untersucht werden können, erscheint unmöglich. Die Wissenschaftler haben daher ein modernes Computeralgebrasystem eingesetzt und die Software RDNets entwickelt, die solche komplizierten Berechnungen innerhalb weniger Minuten durchführt.
Die Forscher untersuchten mit ihrer neuen Software Millionen von Reaktions-Diffusions-Netzwerken und stellten dabei fest, dass die meisten Musterbildungs-Netzwerke eine bisher als absolut notwendig erachtete Bedingung in Turings Modell nicht zwingend erfüllen müssen: Das eine Signalmolekül muss nicht schneller als das andere diffundieren. Die Moleküle können sich auch gleich schnell oder sogar mit beliebigen Geschwindigkeiten bewegen. „Wir haben herausgefunden, dass biologisch relevante Reaktions-Diffusions-Systeme einem völlig anderen Mechanismus folgen als bisher angenommen wurde“, sagt der Forschungsgruppenleiter Patrick Müller.
Die Wissenschaftler haben mit ihrer neuen Software biologische Netzwerke untersucht, die z.B. frühe Differenzierungsvorgänge und die Entwicklung von Fingern steuern. Neben der Relevanz für die Entwicklungsbiologie kann RDNets aber auch von Bioingenieuren verwendet werden, denn mit der Software ist es nun möglich, musterbildende Prozesse zu modellieren und die dazugehörigen genregulatorischen Schaltkreise dann synthetisch nachzubauen. Das ist vor allem für die Gewebezüchtung, beim sogenannten Tissue Engineering, von Nutzen. Die Software kann so dabei helfen, neue synthetische Systeme zu entwicklen, bei denen sich die Zellen an definierten Stellen zu bestimmten unterschiedlichen Gewebearten entwickeln.
Analyse biologischer Netzwerke
Die Systembiologie untersucht biologische Netzwerke, bei denen die Moleküle die Knoten und die molekularen Interaktionen die Kanten der Netzwerke darstellen. Die Dynamik solcher Schaltkreise kann durch Differenzialgleichungen beschrieben werden, die das Verhalten der Moleküle in Abhängigkeit von Ort und Zeit darstellen. Differenzialgleichungen können grundsätzlich auf zwei Arten gelöst werden. Im analytischen Ansatz wird eine Lösung von Hand berechnet, die das Verhalten des Systems für alle möglichen Parameterwerte beschreibt. Im numerischen Ansatz berechnet ein Computer tausende Lösungen, die das Verhalten der Gleichungen unter bestimmten Bedingungen beschreiben, wie zum Beispiel für repräsentative Parameterwerte. Solche numerischen Simulationen können deshalb nur einen Teil des Parameterraums abbilden, wohingegen analytische Ansätze eine vollständige Beschreibung des Systems liefern. In der Praxis sind analytische Ansätze allerdings auf einfache Systeme beschränkt, da die Mathematik mit zunehmender Größe des Systems zu kompliziert wird, um sie von Hand lösen zu können.
Der Ansatz hat diese Einschränkung nun überwunden. Die Wissenschaftler haben das Programm RDNets entwickelt, das eine lineare Stabilitätsanalyse von partiellen Differenzialgleichungen mit Hilfe eines Computeralgebrasystems durchführt. Mit diesem neuartigen Ansatz automatisierter mathematischer Analyse können neue biochemische Turing-Netzwerke gefunden werden, die selbstorganisierende periodische Muster bilden. Die Analyse kann ferner mit qualitativen und quantitativen Bedingungen eingeschränkt werden, sodass die Software den Anwendern völlig neue Möglichkeiten eröffnet, musterbildende Netzwerke während der Embryonalentwicklung zu untersuchen oder synthetische Schaltkreise für Reaktions-Diffusions-Systeme zu entwickeln. RDNets ist frei verfügbar und läuft auf den meisten Internet-Browsern.
IW/HR